-
Towards accurate estimates of the spin-state energetics of spin-crossover complexes within density functional theory: a comparative case study of cobalt(ii) complexes
A. Vargas, I. Krivokapic, A. Hauser and L.M. Lawson Daku
Physical Chemistry Chemical Physics, 15 (11) (2013), p3752-3763


DOI:10.1039/c3cp44336a | unige:26498 | Abstract | Article HTML | Article PDF
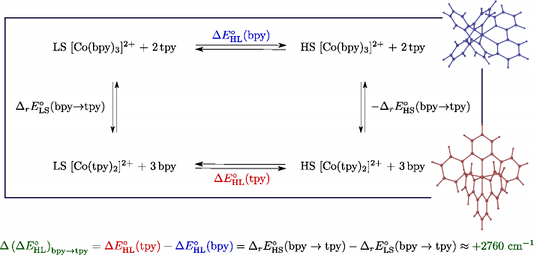
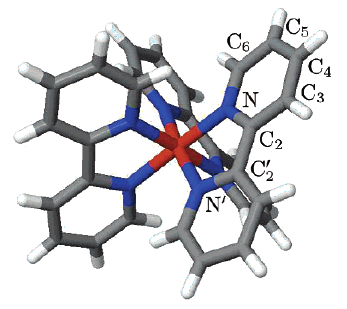
We report a detailed DFT study of the energetic and structural properties of the spin-crossover Co(II) complex [Co(tpy)2]2+ (tpy = 2,2â²:6â²,2â²â²-terpyridine) in the low-spin (LS) and the high-spin (HS) states, using several generalized gradient approximation and hybrid functionals. In either spin-state, the results obtained with the functionals are consistent with one another and in good agreement with available experimental data. Although the different functionals correctly predict the LS state as the electronic ground state of [Co(tpy)2]2+, they give estimates of the HSâLS zero-point energy difference ÎE0HL (tpy)  which strongly depend on the functional used. This dependency on the functional was also reported for the DFT estimates of the zero-point energy difference ÎE0HL (bpy)  in the HS complex [Co(bpy)3]2+ (bpy = 2,2â²-bipyridine) [A. Vargas, A. Hauser and L. M. Lawson Daku, J. Chem. Theory Comput., 2009, 5, 97]. The comparison of the ÎE0HL (tpy)  and ÎE0HL (bpy)  estimates showed that all functionals correctly predict an increase of the zero-point energy difference upon the bpy â tpy ligand substitution, which furthermore weakly depends on the functionals, amounting to (ÎE0HL)bpy->tpy â +2670 cm-1 . From these results and basic thermodynamic considerations, we establish that, despite their limitations, current DFT methods can be applied to the accurate determination of the spin-state energetics of complexes of a transition metal ion, or of these complexes in different environments, provided that the spin-state energetics is accurately known in one case. Thus, making use of the availability of a highly accurate ab initio estimate of the HSâLS energy difference in the complex [Co(NCH)6]2+ [L. M. Lawson Daku, F. Aquilante, T. W. Robinson and A. Hauser, J. Chem. Theory Comput., 2012, 8, 4216], we obtain for [Co(tpy)2]2+ and [Co(bpy)3]2+best estimates of ÎE0HL (bpy) â -2800 cm-1  and ÎE0HL (tpy) â 0 cm-1 , in good agreement with the known magnetic behaviour of the two complexes.